Label the Image to Review the Four Classes of Tissue Grafts
Abstract
The therapeutic potential of donor-derived mesenchymal stromal cells (MSCs) has been investigated in various diseasesane, including steroid-resistant acute graft versus host disease (SR-aGvHD)2. Still, conventional manufacturing approaches are hampered by challenges with scalability and interdonor variability, and clinical trials have shown inconsistent outcomes3,4. Induced pluripotent stem cells (iPSCs) have the potential to overcome these challenges, due to their capacity for multilineage differentiation and indefinite proliferation5,6. Nonetheless, homo clinical trials of iPSC-derived cells have not previously been completed. CYP-001 (iPSC-derived MSCs) is produced using an optimized, good manufacturing practice (GMP)-compliant manufacturing procedure. We conducted a phase 1, open-label clinical trial (no. NCT02923375) in subjects with SR-aGvHD. Xvi subjects were screened and sequentially assigned to cohort A or cohort B (n = viii per group). 1 field of study in cohort B withdrew before receiving CYP-001 and was excluded from assay. All other subjects received intravenous infusions of CYP-001 on days 0 and vii, at a dose level of either ane × 106 cells per kg body weight, to a maximum of 1 × 10viii cells per infusion (cohort A), or two × 10half-dozen cells per kg body weight, to a maximum dose of ii × 108 cells per infusion (cohort B). The master objective was to assess the prophylactic and tolerability of CYP-001, while the secondary objectives were to evaluate efficacy based on the proportion of participants who showed a complete response (CR), overall response (OR) and overall survival (Os) by days 28/100. CYP-001 was safe and well tolerated. No serious adverse events were assessed as related to CYP-001. OR, CR and OS rates by day 100 were 86.7, 53.3 and 86.vii%, respectively. The therapeutic application of iPSC-derived MSCs may now be explored in diverse inflammatory and immune-mediated diseases.
Master
Mesenchymal stromal cells promote an immunosuppressive and immunoregulatory surround by secretion of cytokines, chemokines, growth factors and extracellular vesicles7,8, as well as by activation of indoleamine 2,three-dioxygenase (IDO) product in recipient phagocytes when undergoing apoptosis9. MSCs lack human being leukocyte antigen grade II antigen expression, which allows allogeneic administration without donor–recipient matching. There is general consensus that MSCs derived from primary sources (including bone marrow, adipose tissue, umbilical cord blood and placenta) are condom and well toleratedten.
However, there are substantial scalability and consistency challenges associated with primary donor-derived MSC product. At that place is substantial donor-dependent variability in the propensity of MSCs to exist activated by interferon-gamma and tumor necrosis factor-alpha, which leads to upregulation of IDO expression and results in suppression of T-cell proliferation11,12,thirteen. Additionally, MSC gene expression, differentiation, proliferation and colony-forming chapters vary markedly amongst donors.xiv,15. Furthermore, repeated recruitment and qualification of donors is plush and logistically challenging. While extensive ex vivo culture expansion of MSCs can be employed to produce large numbers of therapeutic doses per donation, this may lead to replicative senescence and other changes to MSC backdrop, even at relatively low culture expansion levels15. Indeed, clinical information suggest that minimally expanded, bone marrow–derived MSCs (BM-MSCs) are associated with ameliorate outcomes in SR-aGvHD patients compared to moderately expanded passage 3–iv BM-MSCs16.
We have developed a new iPSC- and mesenchymoangioblast (MCA)-based manufacturing platform to address these challenges, by eliminating the need to rely on new donors and minimizing the level of civilisation expansion once MSCs are formed.
SR-aGvHD is a disease characterized past very poor prognosis and loftier mortality rates17. Therapeutic use of MSCs for the treatment of SR-aGvHD was introduced past Le Blanc and colleagues in 2004, with positive results18. In a previously published report in a humanized mouse model of aGvHD, the iPSC-derived MSC production CYP-001 reduced bone marrow infiltration and expression of proinflammatory molecules (NOTCH1, TBET and PKCΘ) past CD4 and CD8 T cells, attenuated affliction severity and prolonged survivalxix.
In this written report we have optimized the manufacture of CYP-001 and investigated its safety, tolerability and efficacy for the treatment of adults with SR-aGvHD in a multicenter, phase I, open up-label, dose-escalation clinical trial (NCT02923375). As the first completed human clinical trial of iPSC-derived cells in any disease, this study is relevant not only to SR-aGvHD just also to many other diseases for which MSCs may have therapeutic utility.
The Cymerus proprietary manufacturing process, which is used to manufacture CYP-001, is represented in Fig. one. This new production platform generates MSCs from iPSCs via an apelin receptor+ lateral plate mesoderm intermediate jail cell with fibroblast growth cistron-2 (FGF2)-dependent colony-forming potential, known equally an MCA20,21. This approach allows for the product of well-defined MSCs that express lateral plate, but non paraxial or intermediate, mesoderm markers.

Serum-free medium is used throughout. IMDM, Iscove's modified Dulbecco's medium; F12, Ham'south F12 nutrient mixture; BMP4, bone morphogenic poly peptide-4; ESFM, endothelial serum-free medium.
The manufacturing process consists of three stages: (1) iPSC banking, (2) iPSC expansion and differentiation to MSCs to freeze and intermediate bank and (3) MSC expansion and formulation of final clinical production. Based on the existing banking strategy, approximately 9 × xiv vials, each containing 1 × 106 iPSCs, tin exist generated from a unmarried iPSC line. At the current processing scale, a single vial of one × ten6 iPSCs is capable of giving rise to three.2 × ten10 MSCs on average, while the entire iPSC bank has the capacity to generate ii.9 × ten15 MSCs, or 29 million clinical doses (each containing 1 × 108 MSCs).
The iPSCs used in the manufacture of CYP-001 were derived by reprogramming peripheral blood mononuclear cells from a healthy developed donor using episomal, nonintegrating, oriP/EBNA1−based plasmids.
Nosotros used a PCR analysis on both predifferentiated iPSCs and the finished MCA-derived MSC product to verify that reprogramming plasmids were not integrated into the reprogrammed cells. Comparative genomic hybridization (CGH) and single-nucleotide polymorphism (SNP) assay showed there were no differences in iPSC samples taken before and after expansion through ten passages, indicating that iPSCs are genomically stable during in vitro expansion. The results from the iPSC samples were also compared to an Agilent Reference genome, and there were no mutations of known or potential clinical significance.
We developed an optimized, GMP-compliant manufacturing process utilizing xenogen-, serum- and feeder-gratis conditions. We eliminated the use of murine feeders, in contrast to the original protocol developed by Vodyanik et al.20, instead using chemically defined atmospheric condition for iPSC maintenance as previously described by Chen et al.22. We also optimized the mesenchymal colony-forming medium (M-CFM) compared to that used by Vodyanik et al., and implemented a modified version of the protocol described by Uenishi et al.23 to achieve mesoderm induction. The optimized process generated MSCs with superior functioning in a previously described immunopotency assay24.
To avoid the take a chance of teratoma formation or abnormal differentiation in vivo, the CYP-001 manufacturing procedure was designed to ensure the absenteeism of residual iPSCs in the final production past incorporation of the following steps: (1) afterwards the induction of mesoderm, cells were cultured in a single-jail cell suspension in semisolid medium, which does non support iPSC survival; (2) cells were passed through a mesh filtration pace, which eliminates small clumps of undifferentiated iPSCs; and (three) iPSC-derived MSCs were expanded in adherent jail cell civilisation conditions, which practice not back up survival or expansion of undifferentiated iPSCs.
To confirm these mitigation steps, nosotros conducted an experiment in which undifferentiated iPSCs were seeded in identify of MSC progenitor cells in the M-CFM civilization step. Subsequently culturing for a elapsing equivalent to that of the differentiation process, cells were nerveless and cultured under conditions that support the growth and expansion of human pluripotent stem cells. No iPSC colony formation was observed (lower limit of detection, 0.001%), and microscopic observations detected simply unmarried expressionless cells that did not attach or split in the flowthrough fraction plated. We conclude that residual iPSCs do not survive M-CFM culture, and that any dead iPSCs in this civilization are removed before the MSC expansion stage of the process, providing reassurance that the terminal CYP-001 production is complimentary from residual iPSCs.
The cells in CYP-001 encounter the minimal criteria for defining multipotent MSCs every bit proposed by the International Society for Cell and Gene Therapy19,25. The process is highly efficient, yielding a homogenous population of CD105+, CD73+, CD90+, CD43/45–, CD31– and HLA-DR− MSCs (Supplementary Table 1).
We adult a new assay to quantitate potential remainder undifferentiated stem cells by measuring the LIN28 factor, which is associated with the pluripotent stem cell state. Samples were subjected to a procedure that selectively amplifies the growth of undifferentiated iPSCs in a background of MSCs, to increase the sensitivity of quantitative opposite-transcription polymerase chain reaction (qRT–PCR) for residual iPSC detection (lower limit of detection, 0.001%). We as well performed an in vitro tumorigenicity analysis to observe the potential presence of transformed cells capable of colony germination in soft agar. No contaminating undifferentiated iPSCs or colony formation in soft agar accept been detected in any batch of CYP-001 manufactured to date.
Global gene expression (transcriptome) analysis performed on MCA-derived MSCs demonstrated a very high caste of consistency in gene expression and isoforms between lots (Extended Data Figs. 1 and ii).
Extended Data Fig. iii shows a Consort diagram for the stage I clinical trial. The study recruited male person and female subjects who had received an allogeneic hematopoietic stem prison cell transplant and afterward been diagnosed with grade Two–Iv aGvHD. Subjects were required to meet all of the following criteria: (ane) failure to reply/disease progression after at least iii days' intravenous or oral treatment with an appropriate corticosteroid at a dose of at to the lowest degree 1 mg kg–i d–1; (two) treated with a steroid regimen and elapsing consistent with normal practice at the relevant clinical site; and (3) considered to exist steroid resistant in the opinion of the investigator. These criteria were selected to permit for intersite variability in clinical exercise.
The master objective was to assess the safety and tolerability of CYP-001, while the secondary objective was to evaluate the efficacy of CYP-001 (assessed by best response to treatment by days 28 and 100 and OS at days 28 and 100; Supplementary Table 2). Subjects who completed the 100-day primary evaluation period then entered a 2-year safe and survival follow-up phase.
Betwixt 10 May 2017 and 22 May 2018, a total of sixteen subjects were screened, all of whom were enrolled. Before enrollment, 10 subjects had been treated with corticosteroid doses of 1 mg kg–1 d–1 while six had received doses of 1.v–ii.0 mg kg–one d–ane. Fifteen subjects were treated with CYP-001 in one of ii dose cohorts. One subject withdrew before receiving CYP-001 after experiencing a myocardial infarction, and was therefore excluded from assay. Patients were sequentially assigned to cohort A or cohort B. Subjects enrolled in accomplice A (north = 8) received two doses of CYP-001 (1 × 10six cells per kg body weight upwards to a maximum dose of 1 × 10viii cells, administered intravenously on days 0 and 7). Subjects enrolled in cohort B (north = 7) were treated on the same schedule, using ii × 106 cells per kg torso weight for each dose upwardly to a maximum dose of 2 × x8 cells. Participants received CYP-001 from a full of four different batches in this report.
Baseline characteristics of subjects in the ii cohorts are shown in Supplementary Table three, while baseline aGvHD staging and grading information by subject are shown in Supplementary Table 4.
Handling with CYP-001 was well tolerated, with no notable differences observed between the 2 cohorts (Table 1). Five adverse events (AEs) were assessed by the investigator as perhaps related (defined every bit AEs with a reasonable time relationship to CYP-001 administration, but which could also be explained by disease or other drugs): abdominal hurting, diarrhea, febrile neutropenia, arthralgia and renal impairment (each n = i). No AEs were assessed by the investigator every bit probably or definitely related to CYP-001 treatment. A modest number of AEs (8/104 events) were classified equally severe: febrile neutropenia (due north = 3), cicatrice (northward = two), rash erythematosus (n = one), pruritis (north = 1) and pneumonia (n = 1). One instance of severe febrile neutropenia was assessed by the investigator every bit perhaps related to CYP-001 treatment, and all other severe AEs were assessed as unlikely related or not related. Five serious adverse events (SAEs) were reported (lower respiratory tract infection, pneumonia, delirious neutropenia, parainfluenza virus infection and hypokalemia), merely no SAEs were assessed by the investigator as perchance, probably or definitely related to CYP-001. No subjects discontinued treatment due to AEs.
Of the fifteen subjects enrolled, xiii survived until 24-hour interval 100. One bailiwick in accomplice A died of pneumonia on day 28 (which was deemed unrelated to the report drug) and one subject area in accomplice B showed no comeback in aGvHD and withdrew to embark palliative care on day 22.
A summary of aGvHD responses, OS and aGvHD grade past days 28 and 100 of the chief evaluation menstruation is provided in Table 2, while modify in aGvHD status and best response for individual subjects past day 100 in the main evaluation period is shown in Fig. two. Past day 100, CR and OS were observed in 53.three and 86.7% of subjects, respectively, with no dose-dependent differences. The median time to both starting time response and all-time response was earlier in the high-dose cohort (iii and 14 d, respectively) than in the depression-dose cohort (14 and 60 d, respectively).
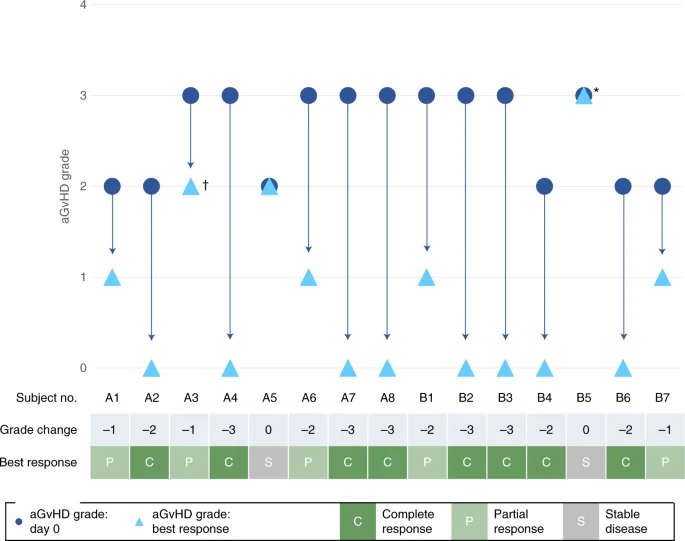
Change in aGvHD status and all-time response in individual subjects by day100. Asterisk (*) shows subject B5 withdrew from the trial on 24-hour interval 22 to commence palliative care; cantankerous (†) shows that subject A3 showed PR at days xiv and 21 simply died due to pneumonia on day 28. Viii subjects were enrolled in each cohort, simply one subject in cohort B withdrew before infusion of CYP-001.
Concomitant aGvHD medication use is summarized in Supplementary Tabular array v. All subjects in both cohorts continued handling with concomitant standard of care aGvHD medications, with universal systemic corticosteroid (glucocorticoid) use. Additional immunosuppressants were used in ten/fifteen subjects (66.7%). Amid the latter, calcineurin inhibitors were prescribed more ofttimes in the low-dose (75%) than in the high-dose (57.one%) cohort, as were selective immunosuppressants (75 versus 14.3%, respectively). Of note, all thirteen subjects who had a solar day 100 visit were either receiving no systemic steroids by that time (n = seven) or receiving a lower dose than at day 0 (n = 6). Additionally, 2 subjects who were on intravenous steroids at mean solar day 0 were transitioned to oral steroids at day 100.
Give-and-take
Our study demonstrates that iPSC-derived MSCs can exist manufactured using a new cell differentiation and expansion platform that eliminates major bug of supply, scalability and consistency.
Our iPSC-based arroyo has the potential to overcome the fundamental limitations of conventional, donor-derived MSC product processes, as information technology facilitates the manufacture of an effectively limitless number of MSCs from a single blood donation. Information technology also avoids the demand for excessive culture expansion of differentiated MSCs, by instead harnessing the indefinite replication potential of iPSCs. Performing MSC product under xenogen-, serum- and feeder-complimentary conditions minimizes the potential for contagion with zoonotic agents and further reduces potential sources of variability. The use of MCA colony-forming mesodermal progenitors allows for the production of a consistent MSC production of well-defined origin. Moreover, the colony-forming step during manufacture provides an additional line of safe since undifferentiated iPSCs are not capable of surviving in MCA-specific, semisolid, colony-forming cultures.
Other groups have explored diverse approaches to overcome the scalability and consistency challenges associated with conventional MSC product. For example, the MSC-FFM method achieves dose-to-dose consistency inside an MSC banking concern derived from pooled bone marrow mononuclear cells from multiple donors, with encouraging clinical trial results26. This facilitates the production of a larger quantity of MSCs from a single bank than a single-donor arroyo, with a similar level of culture expansion. Nevertheless, each bank would suffice for the treatment of simply ~175 patients at the highest dose regimen used in the clinical trial (four infusions at ii × 10half dozen cells per kg body weight). Evidence of consistency between banks produced using this method requires further exploration.
A single iPSC depository financial institution is sufficient to generate ~375,000 batches of CYP-001, equating to approximately 29 million clinical doses at the current processing scale. Our process too has the potential to be scaled up further to produce a considerably higher number of doses per bank every bit development progresses.
Our clinical trial demonstrates that, within a limited number of subjects with SR-aGVHD, CYP-001 is prophylactic and tolerable. Additionally, although further trials with larger sample sizes will be required to confirm efficacy, the aGvHD response and Bone rates observed are encouraging.
Outcomes in our study compare favorably with those in previously published studies involving BM-MSCs and other second-line agents. Yet, such comparisons should be interpreted with caution, in light of differences between trials including sample sizes, the definition of steroid resistance and the use of concomitant medications.
Beyond xv published clinical trials involving the treatment of SR-aGvHD with BM-MSCs2, twenty-four hours 28 OR rates ranged from 45 to 86.5% compared to 62.v% (low dose) and 87.five% (high dose) in our study. Day 28 CR rates later on BM-MSC handling in the aforementioned published trials ranged from 6.5 to 65% compared to 12.5% (depression dose) and 57.i% (high dose) in our study. Day 100 survival rates ranged from 34.four to 57.three% in the iii BM-MSC studies reviewed that reported 100-day survival, compared to 87.5% (low dose) and 85.7% (loftier dose) in our study. Similarly, in a recently published phase Three study, 260 subjects with SR-aGvHD received standard of care plus either (1) placebo or (two) 8 to 12 infusions of BM-MSCs27. The day 28 OR rate in the BM-MSC group was 58%, with a 100-day Os charge per unit of <l%27. Information technology is besides noteworthy that subjects in our study received two infusions each, compared to betwixt 3 and 12 infusions each in the BM-MSC studies.
Other second-line agents used in patients with SR-aGvHD include ruxolitinib, etanercept and extracorporeal photopheresis (ECP). In a recently published phase III report, patients treated with ruxolitinib showed day 28 CR and OR rates of 34 and 62%, respectively28, while response rates observed in a stage 2 report with the same amanuensis were marginally lower29. Day 28 CR and OR rates of 0–20 and fifty–53%, respectively, were reported in trials with etanercept30,31, while in a case series of patients treated with ECP the one-month CR and OR rates were 33 and <50%, respectively32. It is also of import to consider the safe concerns associated with other agents, in particular ruxolitinib and etanercept, in comparing to MSCs.
Based on twenty-four hours 28 responses rates, it appears that the college dose level of CYP-001 led to improved outcomes compared to the lower dose. All the same, we note that patients who received the higher dose were approximately 20 years younger on boilerplate, which may have impacted treatment response, and both groups achieved comparable response rates by day 100.
All subjects were administered corticosteroids, and most (10/15) were administered immunosuppressants during our study. The utilise of other second-line agents after day 28, as was allowed per protocol, may be a further misreckoning gene regarding twenty-four hour period 100 outcomes. Additionally, although we demonstrated that CYP-001 exhibits a highly consistent cistron expression profile between batches, differences in epigenetic and functional features may exist. However, these data highlight the importance of pursuing investigations of iPSC-derived MSCs in the handling of SR-aGvHD and will inform dosing regimens in future studies involving CYP-001.
Although initially tested in aGVHD, iPSC-derived MSCs may be used in the future for a range of other clinical targets. Encouraging information on the potential utility of iPSC-derived MSCs manufactured using this platform have already been generated in preclinical models of critical limb ischemia33, asthma34,35, organ transplant rejection36 and acute respiratory distress syndrome37. Furthermore, nosotros note that primary MSCs have been investigated for a wide range of other therapeutic indications, and nosotros hypothesize that iPSC-derived MSCs could exist expected to show like in vivo effects more generally. The scalability and consistency of this iPSC-derived process could prove to be even more than advantageous if the cells are shown to accept beneficial effects in one or more atmospheric condition with a much higher incidence than aGvHD.
Other studies are investigating diverse iPSC-derived cellular products in conditions including age-related macular degeneration38, heart failure39, Parkinson's disease40 and solid tumors (no. NCT03841110), but clinical trial results have all the same to be reported.
The challenges of scalability, reproducibility and consistency of iPSC-derived products, which have been overcome herein, are pertinent to the application of iPSC technology regardless of the illness target. This study represents the first report of a completed human clinical trial using iPSC-derived cells in whatsoever disease.
Methods
Manufacture of CYP-001
CYP-001 was manufactured on behalf of the sponsor (Cynata Therapeutics Express) by Waisman Biomanufacturing.
The iPSCs used in the manufacture of CYP-001 were derived by Cellular Dynamics International, Inc., by reprogramming peripheral blood mononuclear cells from a healthy adult donor using episomal, nonintegrating, oriP/EBNA1-based plasmids, every bit previously described41.
Donor starting material (whole claret) was collected from healthy adult donors by BloodCenter of Wisconsin, Inc. Donor screening criteria included the standard criteria for altruistic claret, in addition to a number of additional exclusion criteria (provision of informed consent; at least eighteen years old; weight at least 110 pounds (equivalent to 49.9 kg); hemoglobin ≥12.5 g dl–1; no history of bone marrow transplant, cancer or neurological disease). Any bailiwick who met whatever exclusion criterion was automatically excluded from donation. The medical history of prospective donors was reviewed past medical staff to determine whether the bailiwick was suitable to donate, and a class was completed by the master investigator in all cases to confirm eligibility. Additionally, prospective donors were subjected to the standard communicable diseases testing for blood donors. Donor testing and medical examination were performed in compliance with the donor eligibility requirements specified in the US Food & Drug Assistants (FDA) Human being Cells, Tissues, and Cellular and Tissue-Based Products (HCT/P) regulations (no. 21 CFR 1271).
Whole claret samples were nerveless from each donor using standard 250-ml blood bags. One sample was provided for reprogramming while an additional sample was taken for donor testing. All pathogen testing was performed using test kits canonical past the US FDA in a FDA-registered HCT/P testing facility.
Reagents and media used in the manufacture of CYP-001 are described in Supplementary Tables 6–eleven. The process involved the following steps.
- (ane)
iPSCs were thawed in E8 Complete Medium (E8CM; DMEM/F12 Base Medium + E8 Supplement) + 1 µM H1152 (a rho-associated, coiled-curlicue-containing protein kinase (ROCK) inhibitor) on Vitronectin-coated (0.5 µg cm–ii) vi-well plates and incubated at 37 °C, 5% CO2, 20% O2 (normoxia).
- (2)
iPSCs were expanded over three passages in E8CM (without Rock inhibitor) on Vitronectin-coated (0.5 µg cm–two) six-well plates and incubated at 37 °C, 5% COii, 20% O2 (normoxia) before initiation of the differentiation process.
- (iii)
iPSCs were harvested and seeded as single cells/small colonies at 5 × 103 cells cm–2 on Collagen IV-coated (0.v µg cm–2) six-well plates in E8CM + x µM Y27632 and incubated at 37 °C, 5% CO2, 20% O2 (normoxia) for 24 h.
- (4)
E8CM + 10 µM Y27632 was replaced with differentiation medium (Supplementary Tabular array ix) and the plates were further incubated at 37 °C, five% COii, 5% Oii (hypoxia) for 48 h.
- (5)
Colony-forming cells were harvested from differentiation medium-adherent civilisation as a single-cell pause, transferred to M-CFM (Supplementary Tabular array ten) intermission culture in ultra-depression-attachment half-dozen-well plates and incubated at 37 °C, 5% CO2, 20% O2 (normoxia) for 12 d.
- (6)
Colonies (passage 0) were harvested and seeded on Fibronectin/Collagen I-coated (0.67 µg cm–2 Fibronectin, 1.two µg cm–2 Collagen I) plasticware with a modified smoothed finite element method (M-SFEM) and incubated at 37 °C, 5% CO2, 20% O2 (normoxia) for 3 d.
- (7)
From passage ane to 5, colonies were harvested and seeded as single cells at 1.3 × x4 cells cm–ii in G-SFEM on Fibronectin/Collagen 1-coated plasticware and incubated at 37 °C, 5% CO2, xx% Oii (normoxia) for 3 d.
- (8)
The final harvested MCA-derived MSCs were washed with DPBS and harvested into resuspension medium (Plasma-Lyte A Injection pH 7.4 (Baxter) and 10% homo serum albumin (HSA, Baxter)), pooled and centrifuged. A cell count was performed and a final dilution made to achieve a target cell concentration of x × 106 cells ml–1. These cells were then diluted 1:1 with cold cryopreservation solution (Plasma-Lyte A Injection pH 7.4, 10% HSA, 5% DMSO) to requite a terminal cell concentration of 5 × 106 cells ml–1 in Plasma-Lyte A Injection pH 7.4, x% HSA and 2.v% DMSO. Units of the finished product were so filled into CryoStore Freezing Bags (Origen; 20 ml make full volume), immediately cryopreserved in a controlled-rate freezer programmed to a freezing profile of −1 °C min–1 and later stored in the vapor phase of liquid nitrogen.
To confirm the disability of rest iPSCs to survive the K-CFM process stage, we conducted an experiment in which undifferentiated iPSCs were seeded in place of MSC progenitor cells in the M-CFM culture step. Nosotros investigated whether iPSCs could survive in M-CFM civilization. iPSCs were thawed and expanded according to the same procedure used in the manufacture of CYP-001. K-CFM civilization was and then executed as per the CYP-001 process, with the exception that the differentiation procedure was omitted and thus undifferentiated iPSCs were seeded in place of MSC progenitor cells. Afterward culturing for a elapsing equivalent to that of the normal differentiation process, cells were strained through 100-µm strainers and both flowthrough and strained fractions were collected, centrifuged, pooled and resuspended in E8CM plus a Rock inhibitor and cultured in 1 well each of a six-well plate coated with Laminin 521 and E-Cadherin. E8CM is a xenogen- and feeder-gratuitous medium specially formulated for the growth and expansion of human pluripotent stem cells. The Stone inhibitor enhances survival and expansion of singularized iPSCs, while Laminin 521 and E-Cadherin form a coating matrix that also enhances survival and expansion of singularized iPSCs. Collectively, this culture organisation supports iPSC survival and growth, which ways that if any residual undifferentiated iPSCs survive M-CFM culture, they would be expected to survive and proliferate when transferred to this civilisation arrangement.
Quality control
Release tests were performed to ascertain the quality and safety of the manufactured CYP-001 product. A PCR analysis with primers specific to the reprogramming vector used to generate the iPSC line was undertaken to verify that reprogramming plasmids were non integrated into the reprogrammed cells. This assay, which has a lower limit of detection of less than 1 re-create of plasmid per assay, was performed on samples of iPSCs (earlier differentiation) and again on the finished MCA-derived MSC product.
The remainder undifferentiated iPSC assay was performed to quantitate rest undifferentiated stalk cells by TaqMan Gene Expression Analysis (qRT–PCR) targeting LIN28, a gene associated with the pluripotent stem jail cell land. Test samples were subjected to a procedure that selectively amplifies the growth of undifferentiated iPSCs in a background of MSCs, to increase the sensitivity of qRT–PCR for residual iPSC detection. This pace involves culturing the product in E8CM with a ROCK inhibitor on Laminin 521/E-Cadherin-coated cultureware for 6 d.
Cells were prepared for total RNA isolation using RNAprotect Cell Reagent, then centrifuged for 5 min at iv,000 r.p.one thousand. (Sorvall ID 0018 or equivalent) to collect whatsoever formed cells and precipitate. Total RNA was isolated using the QIAGEN RNeasy Plus Mini Kit, complementary Dna was prepared using the Loftier Chapters RNA-to-cDNA Kit and TaqMan Factor Expression Assay was performed for LIN28 and GAPDH. The average cycle where the fluorescent signal crosses the threshold for real-time qPCR and qRT–PCR assays (Cq(50)) and percentage relative southward.d. were calculated for each LIN28 and GAPDH ± the real-time dataset. The boilerplate Cq(50) values were normalized past subtracting the average GAPDH Cq(l) (n = three) from the average LIN28 Cq(fifty) (n = three). Comparisons were performed between samples for normalized average Cq(fifty) values to determine whether the 0.001% iPSC spiked sample (amplified) could exist detected above background and whether GAPDH Cq(50) percentage relative s.d. for all samples was ≤five%.
The in vitro tumorigenicity assay using soft agar colony germination42 was performed as follows. Cells from the examination sample plus a positive control (HT-1080 cells) and a negative command (WI-38) were plated at concentrations of both 1 × 105 and 1 × 106 cells per Petri dish and incubated for 14 d. Photographs were taken of each dish after 14 d to ostend colony formation in the positive control, no colony formation in the negative control and to define relative colony growth in the test sample (at both cell concentrations for each).
Comparative genomic hybridization and SNP analysis were performed to determine whether there was a trend for iPSCs to acquire abnormalities during civilisation expansion. CGH/SNP testing was performed using the Agilent SurePrint G3 Human being CGH + SNP Microarray Kit (four×180k), with a probe length of 60 mer, and analyzed using Agilent CytoGenomics Edition four.0.2.21 software. iPSC samples at passage 21 (the master jail cell banking company passage level) and passage 31 (beyond the passage level used in MCA-derived MSC industry) were tested.
Global gene expression (transcriptome) analysis was performed past messenger RNA sequencing (mRNA-seq) on MCA-derived MSCs produced in 3 separate lots, to decide consistency in factor expression between lots. Full RNA was extracted from MCA-derived MSC samples, from which mRNA was then isolated and analyzed using the Illumina HiSeq 2500 platform. Normalized transcripts per kilobase meg counts for each sample were then calculated and plotted against one another, and the Pearson correlation was computed to decide the degree of consistency of gene expression and isoforms between samples.
Phase I clinical trial of iPSC-derived cells: CYP-001 in astute SR-aGvHD
Details of the study blueprint are provided in the Life Sciences Reporting Summary.
This phase I, multicenter, open up-label, dose-escalation written report investigated the rubber, tolerability and efficacy of ii doses of CYP-001. CYP-001 was supplied as one × ten8 (100 meg) MCA-derived MSCs formulated in 20 ml of serum-complimentary cryoprotectant medium, which contained 2.five% DMSO and 10% HSA. The production was stored and shipped at or below −140 °C and thawed to 37 °C at the clinical site immediately before bailiwick administration.
Subjects were recruited from the Uk (5 sites) and Australia (2 sites) from 10 May 2017 to 28 August 2018. Written informed consent was obtained from all subjects by the primary investigator (or subinvestigator) following full disclosure of the study and before initiation of any study-related assessment or investigation. The study was designed, implemented and reported in accordance with the International Briefing on Harmonization/Harmonized Tripartite Guidelines for Skilful Clinical Practise, with applicable local regulations, and with the ethical principles laid down in the Annunciation of Helsinki. The protocol was canonical by the North East–York Research Ethics Committee, Jarrow, UK, on behalf of all participating centers in the United Kingdom (reference no. 16/NE/0316) and the Purple Adelaide Hospital Homo Research Ethics Committee, Adelaide, Australia, on behalf of both participating centers in Australia (reference no. HREC/16/RAH/412).
Five protocol amendments were made during the elapsing of the study, none of which had a material impact on clinical outcomes. Protocol amendments 1–3 were fabricated earlier written report commencement (6 July 2016, vii November 2016, 22 Nov 2016). Protocol amendment 4 was fabricated on xviii May 2017 (with one subject enrolled into accomplice A) to replace the exclusion criterion of sepsis with severe or uncontrolled systemic infection (bacterial, viral or fungal) likely to impact the subject area's power to participate in the trial. Protocol amendment 5 was made on thirteen Nov 2017 (with seven subjects enrolled into accomplice A) to redefine SR-aGvHD as failure to respond or progress afterward oral treatment in addition to intravenous treatment, and to remove the exclusion criterion relating to Eastern Cooperative Oncology Group grade three or higher.
Study subjects
The report recruited male person and female subjects aged 18–70 years (inclusive) who had undergone an allogeneic hematopoietic stem cell transplant to treat a hematological disorder (including, just non limited to, hematological malignancy) and who had subsequently been diagnosed using consensus grading with grade Two–Iv SR-aGvHD (based on the 1994 Consensus Briefing on Acute GVHD Grading)43.
Prospective participants were required to provide written informed consent earlier screening. All subjects were required to take failed to respond to at least three d of steroid treatment (≥i mg kg–1 d–ane), administered in accordance with standard management at each eye. Specifically, (1) the participant must have failed to reply, or progressed, after at to the lowest degree 3 d of intravenous or oral treatment with an appropriate corticosteroid at a dose of at to the lowest degree 1 mg kg–ane d–1; (2) the steroid regimen and duration administered must be consistent with normal practice at the relevant clinical site; and (3) the participant must accept steroid-resistant aGvHD in the opinion of the investigator. The life expectancy of subjects must have been at least 1 month at the time of screening in the opinion of the investigator. Female participants of childbearing potential were required to render a negative urine pregnancy exam at screening. Participants were as well required to agree to comply with measures intended to prevent female participants becoming significant and male participants fathering children for 3 months following the last dose of CYP-001.
Prospective participants were ineligible if they were: significant or breastfeeding; had received whatever investigational enquiry agent within 30 d or five one-half-lives (whichever is longer) earlier the first dose of CYP-001; had a known or suspected electric current alcohol or substance abuse problem; had a progressive or relapsing hematological malignancy, a current solid tumor or previous cancerous solid tumor probable to recur during the period of the study (except basal or squamous cell carcinomas); had centre failure (New York Heart Association functional class II–Iv) and/or pulmonary failure; were hemodynamically unstable and/or at high take a chance of cardiovascular events; had concluding organ failure (minimal hepatic and/or renal function); had meningitis, pneumonia with hypoxemia, homo immunodeficiency virus or another severe or uncontrolled systemic infection likely to impact on the ability of the patient to participate in the trial; and/or had any other medical or psychiatric condition that made the patient unsuitable for participation in the study.
Written report design, treatments, visits, assessments and outcome measures
Eligible subjects were recruited to either accomplice A (low-dose CYP-001: intravenous infusion of 1 × ten6 cells per kg body weight, upward to a maximum dose of 1 × 10viii cells on days 0 and 7) or cohort B (high-dose CYP-001; intravenous infusion of 2 × 106 cells per kg trunk weight, upwardly to a maximum dose of two × 10eight cells on days 0 and 7) according to a sequential, watch dosing protocol. During the primary evaluation period (days 0–100), subjects were asked to attend study visits on days 0, 3, vii, 14, 21, 28, 60 and 100.
The kickoff viii subjects were enrolled into cohort A. Later the eighth field of study had completed their day 28 assessments, safety data were reviewed by an independent data safety monitoring board to determine whether an additional eight subjects could exist enrolled into cohort B. Infusions were administered using a Fresenius Kabi Volumat MC Agilia Infusion Pump and Fresenius Kabi VL SP22, at a rate of 1 ml min–1. Subjects were also permitted to receive standard of intendance medications. Following completion of the primary evaluation period, surviving subjects then entered a two-year safety and survival follow-up phase.
Issue measures
The chief outcome was safe. In the principal evaluation flow, safety endpoints were based on physical examination, vital signs, pulse oximetry (oxygen saturation), safety laboratory tests (biochemistry, hematology, urinalysis, viral screening), AEs and SAEs. The incidence and severity of AEs up to mean solar day 28 and SAEs possibly related to the written report drug subsequently day 28 were recorded. In the follow-upwardly menstruation, safe endpoints were limited to SAEs accounted possibly, probably or definitely related to the study drug, and malignancy condition.
Efficacy assessments included aGvHD stage/form (according to the 1994 Consensus Conference on Acute GvHD Grading Criteria29; Supplementary Table 2), malignancy status, response condition (CR, partial response (PR), stable disease, disease progression) and survival condition. In the primary evaluation flow, efficacy endpoints were the proportion of subjects showing CR by day 28 and PR past day 28 (both obtained from the 'best' response reported on days iii, seven, xiv, 21 or 28), the proportion of subjects showing CR past 24-hour interval 100 and PR by mean solar day 100 (both obtained from the best response reported on days 3, vii, 14, 21, 28, threescore or 100) and OS at days 28 and 100. In the follow-up catamenia, efficacy endpoints were Bone and aGvHD status at months 6,12,18 and 24, and additional aGvHD treatment required.
Additional assessments included aGvHD treatment received. Blood was also collected for potential time to come biomarker analysis.
Statistical analyses
No formal sample size was calculated for this early phase I safety study, because a accomplice of eight subjects per group is generally accepted as appropriate for testing of initial clinical safety. All prophylactic and efficacy analyses were performed on the safety gear up consisting of all enrolled subjects who received at least i dose of the investigational product, CYP-001, regardless of whether they received the second dose. The principal analysis was performed at the conclusion of the primary evaluation period. Categorical data were summarized using counts and percentages. Continuous data were summarized using mean, median, s.d., minimum and maximum. All data from subjects who withdrew prematurely from the study were included in any analysis where possible. No imputations were made for analysis purposes, and all available information were used in the data summaries. Safety and efficacy results are presented for the safety set from the primary evaluation catamenia only for those subjects who were included in the study for a minimum of 6 months later CYP-001 treatment.
Reporting Summary
Further data on research blueprint is available in the Nature Research Reporting Summary linked to this article.
Information availability
All reasonable requests for raw and analyzed data that are not included in this manuscript or online content will be promptly reviewed past the senior authors to decide whether the request is subject to any intellectual property or confidentiality obligations. Patient-related information may be subject to patient confidentiality restrictions. Whatsoever information and materials that tin can be shared will be released via a cloth transfer agreement. All raw and analyzed global gene expression data tin can exist found at the NCBI Gene Expression Archive (accession no. GSE150969).
References
-
Kabat, M., Bobkov, I., Kumar, Due south. & Grumet, M. Trends in mesenchymal stem jail cell clinical trials 2004–2018: is efficacy optimal in a narrow dose range? Stem Cells Transl. Med. 9, 17–27 (2020).
-
Elgaz, S. et al. Clinical use of mesenchymal stromal cells in the treatment of astute graft-versus-host illness. Transfus. Med. Hemother. 46, 27–34 (2019).
-
Galipeau, J. The mesenchymal stromal cells dilemma—does a negative phase 3 trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy xv, 2–8 (2013).
-
François, M. et al. Human MSC suppression correlates with cytokine induction of indoleamine 2,iii-dioxygenase and bystander M2 macrophage differentiation. Mol. Ther. 20, 187–195 (2012).
-
Takahashi, K. et al. Induction of pluripotent stalk cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
-
Yu, J. et al. Induced pluripotent stem cell lines derived from homo somatic cells. Science 318, 1917–1920 (2007).
-
Galipeau, J. & Sensébé, L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Prison cell 22, 824–833 (2018).
-
Harrell, C. R. et al. Mesenchymal stem cell–derived exosomes and other extracellular vesicles as new remedies in the therapy of inflammatory diseases. Cells 8, 1605 (2019).
-
Galleu, A. et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl. Med. 9, 7828 (2017).
-
Sharma, R. R. et al. Mesenchymal stalk or stromal cells: a review of clinical applications and manufacturing practices. Transfusion 54, 1418–1437 (2014).
-
Wegmeyer, H. et al. Mesenchymal stromal cell characteristics vary depending on their origin. Stem Cells Dev. 22, 2606–2618 (2013).
-
Martin, I. et al. Challenges for mesenchymal stromal jail cell therapies. Sci. Transl. Med. 20, eaat2189 (2019).
-
Ketterl, N. et al. A robust say-so analysis highlights significant donor variation of human mesenchymal stem/progenitor prison cell allowed modulatory capacity and extended radio-resistance. Stalk Jail cell Res. Ther. 6, 236 (2015).
-
Siegel, G. et al. Phenotype, donor age and gender affect function of human bone marrow-derived mesenchymal stromal cells. BMC Med. eleven, 146 (2013).
-
Wagner, West. et al. Replicative senescence of mesenchymal stem cells: a continuous and organized process. PLoS 1 5, e2213 (2008).
-
von Bahr, L. et al. Long-term complications, immunologic effects, and role of passage for issue in mesenchymal stromal jail cell therapy. Biol. Claret Marrow Transplant. xviii, 557–564 (2012).
-
Garnett, C. et al. Treatment and management of graft-versus-host disease: improving response and survival. Ther. Adv. Hematol. iv, 366–378 (2013).
-
Le Blanc, Yard. et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet 363, 1439–1441 (2004).
-
Ozay, East. I. et al. Cymerus™ iPSC-MSCs significantly prolong survival in a pre-clinical, humanized mouse model of graft-vs-host disease. Stalk Cell Res. 35, 101401 (2019).
-
Vodyanik, Grand. A. et al. A mesoderm-derived precursor for mesenchymal stalk and endothelial cells. Cell Stem Jail cell 7, 718–729 (2010).
-
Slukvin, I. I. & Kumar, A. The mesenchymoangioblast, mesodermal precursor for mesenchymal and endothelial cells. Cell. Mol. Life Sci. 75, 3507–3520 (2018).
-
Chen, G. et al. Chemically divers weather condition for human iPS cell derivation and culture. Nat. Methods viii, 424–429 (2011).
-
Uenishi, G. et al. Tenascin C promotes hematoendothelial development and T lymphoid commitment from human being pluripotent stalk cells in chemically defined weather. Stalk Cell Rep. 3, 1073–1084 (2014).
-
Slukvin, I., Uenishi, M., Hei, D. & Drier, D. Colony forming medium and use thereof. Patent WO2017156580A1 (2017).
-
Dominici, M. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Guild for Cellular Therapy position argument. Cytotherapy 8, 315–317 (2006).
-
Bader, P. et al. Constructive treatment of steroid and therapy-refractory acute graft versus-host illness with a novel mesenchymal stromal prison cell product (MSC-FFM). Bone Marrow Transplant. 53, 852–862 (2018).
-
Kebriaei, P. et al. A phase 3 randomized study of remestemcel-L versus placebo added to second-line therapy in patients with steroid-refractory acute graft-versus-host disease. Biol. Blood Marrow Transplant. 26, 835–844 (2020).
-
Zeiser, R. et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N. Engl. J. Med. 382, 1800–1810 (2020).
-
Jagasia, K. et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label, phase 2 trial. Blood 135, 1739–1749 (2020).
-
Park, J. H. et al. Etanercept for steroid-refractory acute graft versus host disease following allogeneic hematopoietic stem cell transplantation. Korean J. Intern. Med. 29, 630–636 (2014).
-
De Jong, C. N. et al. Etanercept for steroid-refractory acute graft-versus-host disease: a single middle experience. PLoS One 12, e0187184 (2017).
-
Greinix, H. T. et al. Extracorporeal photochemotherapy in the treatment of astringent steroid-refractory acute graft-versus-host disease: a pilot study. Blood 96, 2426–2431 (2000).
-
Koch, J. M. et al. Mesenchymoangioblast-derived mesenchymal stromal cells inhibit jail cell damage, tissue damage and meliorate peripheral blood flow following hindlimb ischemic injury in mice. Cytotherapy 18, 219–228 (2016).
-
Royce, Due south. 1000. et al. Intranasal administration of mesenchymoangioblast-derived mesenchymal stem cells abrogates airway fibrosis and airway hyperresponsiveness associated with chronic allergic airways disease. FASEB J. 31, 4168–4178 (2017).
-
Royce, Southward. G. et al. iPSC- and mesenchymoangioblast-derived mesenchymal stem cells provide greater protection confronting experimental chronic allergic airways illness compared with a clinically used corticosteroid. FASEB J. 33, 6402–6411 (2019).
-
Khan, M. A. et al. iPSC-derived MSC therapy induces immune tolerance and supports long-term graft survival in mouse orthotopic tracheal transplants. Stem Cell Res. Ther. 10, 290 (2019).
-
Millar, J. E. et al. Combined mesenchymal stromal cell therapy and ECMO in ARDS: a controlled experimental written report in sheep. Am. J. Respir. Crit. Care Med. https://doi.org/10.1164/rccm.201911-2143OC (2020).
-
Mandai, M., Kurimoto, Y. & Takahashi, Grand. Autologous induced stem-cell-derived retinal cells for macular degeneration. N. Engl. J. Med. 377, 792–793 (2017).
-
Ylä-Herttuala, Southward. iPSC-derived cardiomyocytes taken to rescue infarcted heart musculus in coronary middle affliction patients. Mol. Ther. 26, 2077 (2018).
-
Stoddard-Bennett, T. & Reijo Pera, R. Treatment of Parkinson's disease through personalized medicine and induced pluripotent stem cells. Cells 8, 26 (2019).
-
Mack, A. A. et al. Generation of induced pluripotent stem cells from CD34+ cells across blood drawn from multiple donors with not-integrating episomal vectors. PLoS Ane 6, e27956 (2011).
-
Borowicz, South. et al. The soft agar colony formation analysis. J. Vis. Exp. 92, e51998 (2014).
-
Przepiorka, D. et al. 1994 Consensus Briefing on Acute GVHD Grading. Os Marrow Transplant. 15, 825–828 (1995).
Acknowledgements
We give thanks the University of Wisconsin Bioinformatics Resources Center for providing mRNA-seq analysis services, and all site investigators, coinvestigators, study coordinators, other study staff and study participants. Medical writer C. Markey, of Markey Medical Consulting Pty Ltd, assisted in the training of an before version of a typhoon manuscript describing this work. J.E.J.R. is supported by a National Wellness and Medical Enquiry Council Investigator Grant (1177305) for 'driving clinical prison cell and gene therapy in Commonwealth of australia', as well as funding from Cure the Futurity, Therapeutic Innovation Australia and an anonymous foundation.
Author data
Affiliations
Contributions
I.S., G.I.U., D.D. and D.H. adult and optimized the manufacturing process. I.S., D.D., D.H. and L.Southward.L. developed and optimized quality control assays and designed the experiment to verify that iPSCs do non survive M-CFM culture. K.K. designed the clinical trial and wrote the protocol. A.J.C.B., A.P., J.E.G., M.H.G., R.R., D.T.Y. and J.East.J.R. were principal investigators in the clinical trial. Thousand.K. and J.East.J.R. prepared the commencement drafts of the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Patent applications (some of which have been granted) covering aspects of the technology described in this manuscript have been filed by Wisconsin Alumni Research Foundation (which invests in the University of Wisconsin-Madison), Cellular Dynamics International (now FUJIFILM Cellular Dynamics) and Cynata Therapeutics. The patents with inventors including D.D., D.H., Grand.I.U., K.K. and I.Due south. cover the processes used to generate iPSCs and differentiate iPSCs into MSCs, as well as the rest undifferentiated iPSC assay. Cynata Therapeutics funded the work described in this manuscript. K.Thousand. is an employee and shareholder of Cynata Therapeutics. I.S. is a cofounder, shareholder and scientific counselor of Cynata Therapeutics. Fifty.S.50. and D.D. are employees of Waisman Biomanufacturing, Academy of Wisconsin-Madison, which provides contract manufacturing services to Cynata Therapeutics. D.H. is a quondam employee of both Waisman Biomanufacturing and Cellular Dynamics International. Thousand.I.U. and J.E.J.R. take received travel grants from Cynata Therapeutics. A.J.C.B., A.P., J.E.Yard., Grand.H.G., R.R. and D.T.Y. declare no relevant competing interests.
Additional data
Peer review information Jerome Staal was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher'due south notation Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Supplementary information
Rights and permissions
Virtually this article

Cite this article
Bloor, A.J.C., Patel, A., Griffin, J.East. et al. Production, prophylactic and efficacy of iPSC-derived mesenchymal stromal cells in acute steroid-resistant graft versus host disease: a phase I, multicenter, open-characterization, dose-escalation report. Nat Med 26, 1720–1725 (2020). https://doi.org/10.1038/s41591-020-1050-ten
-
Received:
-
Accepted:
-
Published:
-
Issue Date:
-
DOI : https://doi.org/x.1038/s41591-020-1050-x
Further reading
Source: https://www.nature.com/articles/s41591-020-1050-x
0 Response to "Label the Image to Review the Four Classes of Tissue Grafts"
Post a Comment